Curso interactivo de Sistemática Filogenética
Tema 6.- Aplicaciones de la Sistemática Filogenética. Introducción a la Filogenia Molecular.
La filogenia molecular consiste en la obtención de información filogenética a partir del estudio de secuencias moleculares, es decir, ácidos nucleicos (DNA, RNA) o proteínas. Vamos a mostrar aquí cómo los métodos de la Sistemática Filogenética tienen una gran aplicación en filogenia molecular. Muchos piensan que la base de la filogenia molecular consiste en la comparación directa de secuencias, de forma que se consideran más relacionados los organismos en la medida en que sus secuencias son similares. Esta idea (que no es totalmente cierta, como luego veremos) tiene sus raíces en dos argumentos:
- Intuitivo. Parece obvio que cuando dos linajes biológicos divergen y evolucionan independientemente sus secuencias macromoleculares acumulan diferencias progresivamente. A más divergencia, más diferencia.
- Histórico. De hecho en el pasado se aplicaron técnicas que cuantificaban el grado de homología entre dos secuencias, y utilizaban dicho índice para establecer relaciones de parentesco entre organismos.
De nuevo nos encontramos en este campo el mismo dilema que el que exponíamos en el tema 1 sobre los fundamentos de la Sistemática, es decir, similitud versus parentesco filogenético. żDebemos considerar como medida de la divergencia filogenética entre dos organismos el grado absoluto de similitud entre las secuencias o debemos hacer un "análisis de caracteres" como el que hemos realizado en anteriores temas? żPodemos distinguir sinapomorfías en los caracteres moleculares? Vamos a aclarar este punto con un ejemplo.
Para simplificar, vamos a considerar el caso de tres organismos en los que compararemos una pequeńa secuencia (homóloga, evidentemente) de cuatro bases:
| Organismo | Secuencia |
| 1 | AGCC |
| 2 | AGCT |
| 3 | GGTT |
Para trabajar con secuencias es frecuente comenzar con un procedimiento que no suele aplicarse en el caso de caracteres morfológicos, el de elaborar un árbol "no enraizado" (unrooted tree). La idea es construir un "árbol" conectando los taxones mediante ramas, pero de forma que las conexiones impliquen el menor número posible de cambios (otra vez el criterio de parsimonia). En el caso de nuestro ejemplo, tenemos que el árbol no enraizado de máxima parsimonia es:
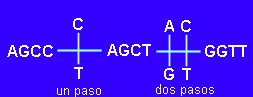 Las barras verticales representan los pasos o cambios, tres en total. De
momento no sabemos en qué dirección se producen dichos cambios, si la sustitución de la
izquierda, en la cuarta posición de la secuencia, es en el sentido C->T o T->C. Lo
que sí sabemos es que cualquier otra alternativa es menos parsimoniosa, por ejemplo:
Las barras verticales representan los pasos o cambios, tres en total. De
momento no sabemos en qué dirección se producen dichos cambios, si la sustitución de la
izquierda, en la cuarta posición de la secuencia, es en el sentido C->T o T->C. Lo
que sí sabemos es que cualquier otra alternativa es menos parsimoniosa, por ejemplo:
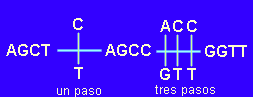
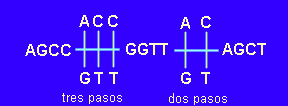
Nos quedamos, por tanto con la primera opción. Pues bien, podemos avanzar más si somos capaces de "enraizar" al árbol, operación posible si conocemos la secuencia de bases en un grupo externo. Esto nos permitiría formular una hipótesis sobre la dirección de los cambios. Supongamos que en el grupo externo elegido la secuencia es AGCC. En este caso ya podemos construir un árbol enraizado que es, en realidad, un cladograma:
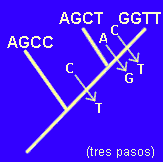 Ahora nos movemos ya por un terreno conocido. En efecto, la
consideración de la secuencia AGCC como ancestral nos ha permitido identificar como
derivados o apomorfos los cambios C->T en la cuarta posición, A->G
en la primera posición y C->T en la tercera posición. Sin embargo, de estos tres
cambios, sólo el primero, la sustitución de la citosina por la timina en la cuarta
posición constituye un carácter derivado y compartido,
es decir, una "sinapomorfía molecular"
que nos permite sugerir una mayor relación de parentesco entre los organismos 2 y 3. En
el fondo el procedimiento es el mismo que con los caracteres morfológicos, aunque la
complejidad del análisis de las secuencias macromoleculares, con cientos o miles de
posiciones, requiere el uso de ordenadores y algoritmos que funcionan de acuerdo con el
razonamiento aquí expuesto. Los programas de análisis filogenético mediante máxima
parsimonia buscan los árboles más cortos (con menos pasos) y efectúan un enraizamiento
con respecto a la secuencia propuesta como ancestral.
Ahora nos movemos ya por un terreno conocido. En efecto, la
consideración de la secuencia AGCC como ancestral nos ha permitido identificar como
derivados o apomorfos los cambios C->T en la cuarta posición, A->G
en la primera posición y C->T en la tercera posición. Sin embargo, de estos tres
cambios, sólo el primero, la sustitución de la citosina por la timina en la cuarta
posición constituye un carácter derivado y compartido,
es decir, una "sinapomorfía molecular"
que nos permite sugerir una mayor relación de parentesco entre los organismos 2 y 3. En
el fondo el procedimiento es el mismo que con los caracteres morfológicos, aunque la
complejidad del análisis de las secuencias macromoleculares, con cientos o miles de
posiciones, requiere el uso de ordenadores y algoritmos que funcionan de acuerdo con el
razonamiento aquí expuesto. Los programas de análisis filogenético mediante máxima
parsimonia buscan los árboles más cortos (con menos pasos) y efectúan un enraizamiento
con respecto a la secuencia propuesta como ancestral.
Es muy importante que en nuestro ejemplo comparemos el resultado final, expresado en el cladograma, con el que habríamos obtenido a partir de la aplicación de un método de distancia (similitud). En efecto, el porcentaje de identidad entre las secuencias es el siguiente:
| AGCT | GGTT | |
| AGCC | 75%(3 de 4) | 25% (1 de 4) |
| GGTT | 50% (2 de 4) |
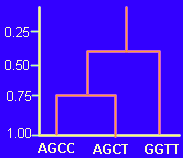
A la derecha tenemos un fenograma (ver tema 1) que representa la similitud (en tanto por uno) mediante el algoritmo UPGMA. Puede comprobarse como la menor distancia se da entre los organismos 1 y 2, que comparten el 75% de la secuencia.
Es decir, ˇlos métodos de distancia hubieran dado un resultado completamente diferente al obtenido por el método de parsimonia! En efecto, un análisis basado en la similitud de las secuencias se hubiera centrado en los caracteres plesiomorfos (no informativos) A y C en la primera y tercera posición. Sin embargo, el único carácter con información filogenética, la T de la cuarta posición, hubiera pasado desapercibida.
Normalmente, el análisis de secuencias no será nunca tan sencillo como en el ejemplo anterior. La homoplasia, es decir, el número total de convergencias y reversiones, será importante, especialmente cuando trabajemos con secuencias de nucleótidos. En este caso, al manejar sólo cuatro letras, la probabilidad de que coincidan de forma independiente, o de que se retorne al estado ancestral, es relativamente alta. Por esto, en filogenia molecular trabajaremos frecuentemente con longitudes de árbol y con índices de consistencia, que nos darán la medida de la "bondad" de las agrupaciones.
